
The stereodynamics of the two reactions: H + LiH+(v = 0, j = 0) → H2 + Li+ and H+ + LiH(v = 0, j = 0) → H2+ + Li
Abstract
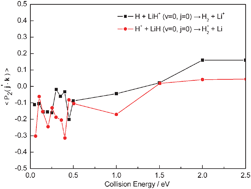
Quasi-classical trajectory (QCT) calculations have been carried out to study the stereodynamics of the reactions H + LiH+ (v = 0, j = 0) → H2 + Li+ and H+ + LiH (v = 0, j = 0) → H2+ + Li which proceed on the two lowest-lying electronic states of the LiH2+ system, using the ab initio potential energy surfaces (PESs) of Martinazzo et al. [J. Chem. Phys., 2003, 119, 11241]. Differential cross sections (DCSs) and alignments of the product rotational angular momentum for the two reactions are reported. Though the two PESs employed in the current calculations have significant differences, the tendencies of the product rotational alignment are same on the whole, and some common features emerge. This interesting phenomenon probably indicates that, for this system, the characters of the PESs have a weak influence on the alignments of the products. The conclusion is confirmed by a further discussion of rotational alignment parameter 〈P2(j′·k)〉 which also indicates that the two PESs are repulsive, i.e., the exoergic processes of the reactions taking place on the exit valleys of the PESs.
 Please wait while we load your content...
Please wait while we load your content...

