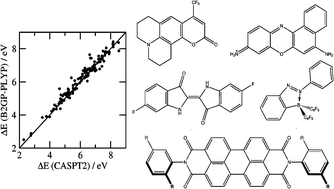
Time-dependent double-hybrid density functional methods are evaluated for the calculation of vertical singlet–singlet valence excitation energies of a wide variety of organic molecules. Beside the already published TD-B2-PLYP method, an analogous approach based on the recently published ground state B2GP-PLYP functional is presented for the first time. Double-hybrid functionals contain a hybrid-GGA-like part for which a conventional TDDFT linear response treatment is carried out. The thus obtained excitation energies are afterwards corrected by adding a non-local correlation portion, which is based on an CIS(D) type excited state perturbative correction. Both, TD-B2-PLYP and TD-B2GP-PLYP, are first applied to the 142 vertical singlet excitation energies in a benchmark set by Schreiber et al., that contains small and medium sized organic molecules. In a second part, a new benchmark set composed of five large organic dyes is proposed. Accurate reference values are derived from experimental 0–0 excitation energies in solution. A back-correction scheme based on TDDFT computations is presented by which solvent, relaxation and vibrational effects are removed, yielding experimental vertical gas phase excitation energies with an estimated accuracy of about ±0.1 eV. The TD-B2-PLYP, TD-B2GP-PLYP and a variety of conventional TDDFT methods are then applied to this new benchmark set. The results for both considered test sets show that the new double-hybrid approaches yield the smallest mean absolute deviations of 0.22 eV for the first benchmark set and 0.19 eV (TD-B2-PLYP) and 0.16 eV (TD-B2GP-PLYP) for the new organic dye test set. Apart from a break-down of the perturbative correction for very high-lying transitions (larger than 8 eV), it is generally found that the double-hybrid functionals show high robustness and accuracy that cannot be obtained with conventional density functionals (e.g. B3-LYP).
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


