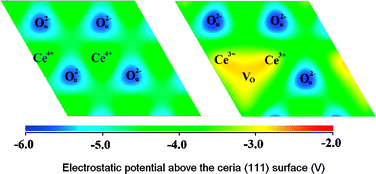
We use density functional theory calculations with Hubbard corrections (DFT+U) to investigate electronic aspects of the interaction between ceria surfaces and gold atoms. Our results show that Au adatoms at the (111) surface of ceria can adopt Au0, Au+ or Au− electronic configurations depending on the adsorption site. The strongest adsorption sites are on top of the surface oxygen and in a bridge position between two surface oxygen atoms, and in both cases charge transfer from the gold atom to one of the Ce cations at the surface is involved. Adsorption at other sites, including the hollow sites of the surface, and an O–Ce bridging site, is weaker and does not involve charge transfer. Adsorption at an oxygen vacancy site is very strong and involves the formation of an Au− anion. We argue that the ability of gold atoms to stabilise oxygen vacancies at the ceria surface by moving into the vacancy site and attracting the excess electrons of the defect could be responsible for the enhanced reducibility of ceria surfaces in the presence of gold. Finally, we rationalise the differences in charge transfer behaviour from site to site in terms of the electrostatic potential at the surface and the coordination of the species.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


