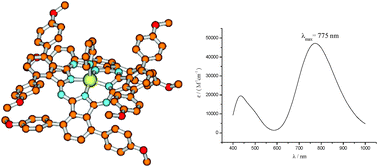
A time-dependent density functional theory study (TD-DFT) is presented regarding the substituent effects on the Q-bands of two classes of non-planar phthalocyanines: the α-octaphenyl and p-α-octamethoxyphenyl substituted compounds, in their free-base and zinc complex forms. Singlet vertical excitation energies, computed at the PBE0/SVP//BP86/SVP level of theory also including bulk solvent effects (COSMO model), resulted within 0.1 eV of experiment. The experimental red-shift for the Q-band, going from the phenylated to the methoxyphenylated case, was well-reproduced theoretically and in the latter case it was found to depend mainly on the nature of the substituents and partly on structure distortion effects. The energetic gap between the singlet ground and first triplet excited state was calculated in solvent to be 1.28 eV for the free-base phthalocyanine (H2Pc) and 1.45 eV for the unsubstituted zinc complex (ZnPc) and lower than 0.98 eV for all the other compounds, which is the energetic lower limit for a molecule to act as photosensitiser in photodynamic therapy according to a Type II reaction mechanism. As a consequence, since this property-requirement for drugs used in photodynamic therapy is not fulfilled by the investigated near-infrared photosensitizers, they cannot be proposed as candidates for their use in this medical treatment.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


