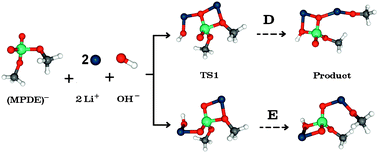
The phosphodiester linkage central to biological systems has been modeled by methyl phosphodiester (MPDE) in various theoretical and experimental studies. Under physiological conditions, hydrolysis of the phosphodiester is negligible, however this process can be catalyzed in the presence of metal ions. To understand the role of alkali metals in MPDE hydrolysis and, in particular, how it influences the reaction pathway and the associated energetics, density functional calculations employing the 6-31+G(d,p) basis set have been carried out. Different pathways that include the reactant, intermediates and the products have been investigated for MPDE hydrolysis catalyzed by one or two lithium ions, characterized as stationary point geometries on the potential energy surface. The pathways A and B incorporate a single lithium ion bonded to different oxygens of the diester functionality. In pathway C, a six-membered ring was noticed wherein the nucleophile bridges two lithium ions interacting with different oxygens of the phosphoryl group. Furthermore, in the pathway (D) incorporating two lithium ions, one of the lithium ions interacts with the hydroxyl group and another with the methoxy oxygen; both metal ions are coordinated by the same phosphoryl oxygen. In addition to this, yet another pathway (E), where the metal ions are bound to different oxygens of the phosphoryl group, has also been dealt with. The calculations have shown that the A and B pathways lead to a single step reaction. A three-step mechanism including the nucleophilic (hydroxyl) attack, rotation of a methyl group and, finally, departure of the methoxy group has been predicted for the D and E profiles. Both D and E pathways are favored equally (with a marginal difference of 0.3 kJ mol−1 in their activation energies) in the gas phase and a transition state corresponding to nucleophilic attack with an energy barrier of 32.5 kJ mol−1 was located when lithium was used. A penta-coordinated phosphorous intermediate on the potential energy surface was characterized along these pathways. MPDE hydrolysis yielded a lower energy barrier for lithium than those for the remaining alkali metal ions. This agrees well with the experimentally observed trend for the hydrolysis rates: Li > Na > K. Self consistent reaction field (SCRF) calculations reveal the lower energy barrier between the reactant and the transition state for the nucleophilic attack in nonpolar solvents. The extent of bond formation (or cleavage) in different stationary point structures along the reaction path as estimated from the electron density at the bond critical point in the molecular electron density topography, has proven useful in distinguishing the associative or dissociative reaction pathways.

 Please wait while we load your content...
Please wait while we load your content...

