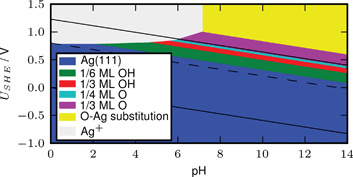
Based on density functional theory calculations we investigate the electrochemically most stable surface structures as a function of pH and electrostatic potential for Pt(111), Ag(111) and Ni(111), and we construct surface Pourbaix diagrams. We study the oxygen reduction reaction (ORR) on the different surface structures and calculate the free energy of the intermediates. We estimate their catalytic activity for ORR by determining the highest potential at which all ORR reaction steps reduce the free energy. We obtain self-consistency in the sense that the surface is stable under the potential at which that particular surface can perform ORR. Using the self consistent surfaces, the activity of the very reactive Ni surface changes dramatically, whereas the activity of the more noble catalysts Pt and Ag remains unchanged. The reason for this difference is the oxidation of the reactive surface. Oxygen absorbed on the surface shifts the reactivity towards the weak binding region, which in turn increases the activity. The oxidation state of the surface and the ORR potential are constant versus the reversible hydrogen electrode (RHE). The dissolution potential in acidic solution, on the other hand, is constant vs. the standard hydrogen electrode (SHE). For Ag, this means that where the potential for dissolution and ORR are about the same at pH = 0, Ag becomes more stable relative to RHE as pH is increased. Hence the pH dependent stability offers an explanation for the possible use of Ag in alkaline fuel cell cathodes.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


