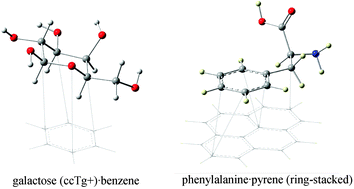
Density functional theory (DFT-D) and semi-empirical (PM3-D) methods having an added dispersion correction have been used to study stabilising carbohydrate–aromatic and amino acid–aromatic interactions. The interaction energy for three simple sugars in different conformations with benzene, all give interaction energies close to 5 kcal mol−1. Our original parameterization of PM3 (PM3-D) seriously overestimates this value, and has prompted a reparametrization which includes a modified core–core interaction term. With two additional parameters, the carbohydrate complexes, as well as the S22 data set, are well reproduced. The new PM3 scheme (PM3-D*) is found to describe the peptide bond–aromatic ring interactions accurately and, together with the DFT-D method, it is used to investigate the interaction of six amino acids with pyrene. Whilst the peptide backbone can adopt both stacked and T-shaped structures in the complexes with similar interaction energies, there is a preference for the unsaturated ring to adopt a stacked structure. Thus, peptides in which the latter interactions are maximised are likely to be the most effective for the functionalisation of carbon nanotubes.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


