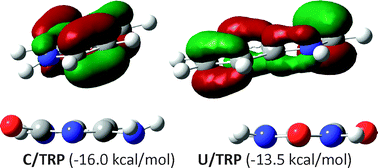
The DF-MP2 quantum chemistry method was applied to a description of the stacking interactions of uracil and cytosine with five model amino acid residues, namely histidine (HIS), phenylalanine (PHE), tyrosine (TYR), tryptophan (TRP) and an arginyl moiety (ARG). The BSSE and complete basis set corrections were taken into account. Both uracil (U) and cytosine (C) may strongly interact with amino acid residues. The stacking energy is very sensitive to both the nature of the interacting monomers and their spatial conformations. However, usually considerable configurational degrees of freedom are observed, leading to similar stacking energies. The overall order of optimized stacking complexes corresponds to the following sequence: C–TRP (−16.0 kcal mol−1) > U–TRP (−13.5 kcal mol−1) > U–TYR (−12.2 kcal mol−1) > U–HIS (−8.7 kcal mol−1) > U–PHE (−7.7 kcal mol−1) > C–PHE (−6.6 kcal mol−1). Cytosine may also strongly attract HIS and TYR via stacking interactions but the corresponding minima were not found since hydrogen-bonded pairs are result of gradient optimizations. Besides, stacking imposes an increase in aromatic character on both analyzed pyrimidines. This is consistently described by changes of both energetic and structural aromaticty indices. There are also observed changes in aromaticities of amino acid residues but the predictions by the harmonic oscillator model of aromaticity (HOMA) index and nucleus-independent chemical shifts (NICS) are inconsistent. Finally, there is also an interesting observation with respect to the extrapolation of the stacking energies: the same quality of complete basis set limits may be obtained without actual calculations on the aug-cc-pVQZ basis set and application of the extrapolation procedure twice gives substantially the same complete basis set results within 0.1 kcal mol−1.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


