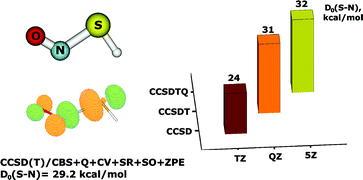
High-level ab initio calculations employing the CCSD and CCSD(T) coupled cluster methods with a series of systematically convergent correlation-consistent basis sets have been performed to obtain accurate molecular geometry and energetic properties of the simplest S-nitrosothiol (RSNO), HSNO. The properties of the S–N bond, which are central to the physiological role of RSNOs in the storage and transport of nitric oxide, are highlighted. Following corrections for quadruple excitations, core-valence correlation and relativistic effects, the CCSD(T) method extrapolated to the complete basis set (CBS) limit yielded values of 1.85 Å and 29.2 kcal mol−1 for the S–N bond length and the dissociation energy for homolysis of the S–N bond, respectively, in the energetically most stable trans-conformer of HSNO. The properties of the S–N bond strongly depend on the basis-set size and the inclusion of triple, and, to a lesser extent, quadruple excitations in the coupled cluster expansion. CCSD calculations systematically underestimate the S–N equilibrium distance and S–N bond dissociation energy by 0.05–0.07 Å and 6–7 kcal mol−1, respectively. The significant differences between the CCSD(T) and CCSD descriptions of HSNO, the high values of the coupled clusterT1 (0.027) and D1 (0.076) diagnostics, as well as the instability of the reference restricted Hartree–Fock (RHF) wavefunction indicate that the electronic structure of the SNO group possesses multireference character. Previous quantum-chemical data on RSNOs are reexamined based on the new insight into the SNO electronic structure obtained from the present high-level calculations on HSNO.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


