
Mechanistic and kinetic study of the O + CH3OCH2 reaction and the unimolecular decomposition of CH3OCH2O†
Abstract
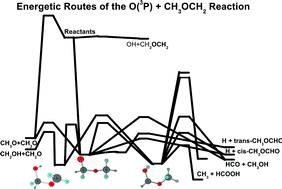
Potential energy surface for the O + CH3OCH2 reaction is calculated using the coupled cluster theory with single, double, and non-iterative triple substitutions [CCSD(T)] with a complete basis set extrapolation. It is revealed that the reaction of O with CH3OCH2 proceeds dominantly via an addition/elimination mechanism. Other minor mechanisms include direct hydrogen abstraction, which may play a significant role at high temperatures, and a high-barrier SN2 displacement. The initial adduct is the CH3OCH2O radical, which has many product channels via
 Please wait while we load your content...
Please wait while we load your content...

