
Ab initio cluster calculations for the absorption energies of F and F+ centers in bulk ZnO
Abstract
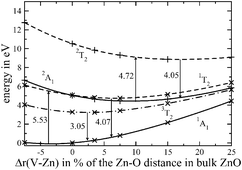
We present the results of a series of ab initio calculations on the ground states and the low lying excited states of the F and F+ centers in bulk ZnO. Both types of F centers are oxygen vacancies, causing rather strong distortions of the local geometries. The calculations were performed by means of wave function based methods, mostly at the CASSCF level. Dynamic correlation was included for the first two coordination shells of the F centers. The calculated absorption energy for the F+ center (3.19 eV) is in excellent agreement with the experimental value of 3.03 eV. For the emission from the 3T2 state of the F center to the 1A1 ground state we obtained a transition energy of 2.73 eV. Experimentally, a green photoluminescence is observed at 2.38–2.45 eV. We estimated that the errors in our calculation should be even smaller in the latter case than for the F+ state, where the calculated transition energy differs by less than 0.2 eV from the experimental value. Therefore, we assume that the 3T2 to 1A1 transition is not the origin of the green luminescence.
 Please wait while we load your content...
Please wait while we load your content...

