
The interplay of non-covalent interactions determining the antiparallel conformation of (isocyanide)gold(i) dimers†

Abstract
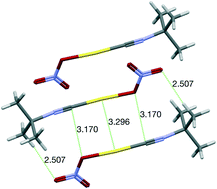
A combined structural and computational analysis of the interaction topology of (isocyanide)gold(I) dimers in the solid state is presented here. After a survey of the Cambridge Structural Database, we have found that, whenever possible, an antiparallel conformation is preferred. An NBO analysis has disclosed the interplay of ligand⋯ligand and ligand⋯gold interactions that accompany the aurophilic contact. We have extended the NBO study to the Au⋯Au interaction in order to try to rationalize it in terms of localized orbitals. The molecular electrostatic potential of the molecules involved has also been mapped. Finally, the destabilization associated with the loss of the antiparallel conformation in the dimer has been analysed by means of long-range dispersion-corrected DFT calculations.
 Please wait while we load your content...
Please wait while we load your content...

