
Thermodynamic parameters of the pedal motion in the crystal structures of two bromomethylated azobenzenes†
Abstract
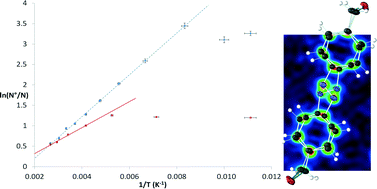
Two bromomethylated azobenzene derivatives were characterized by X-ray crystallography at 100 K after fast cooling, and showed disorder of the central N-atoms. The structures were re-determined over a range of temperatures, providing evidence for dynamic disorder due to pedal motion of the central N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. Using van't Hoff plots, thermodynamic parameters for the pedal motion were determined. Computationally very cheap Atom–Atom-Force Field (AA-CLP) calculations were employed, which showed that the differences in dynamic disorder enthalpy between the two compounds are predominantly due to intermolecular interactions. AA-CLP calculations and gas phase electronic structure calculations were employed to show the link between intermolecular interactions and activation energy for pedal motion.
N bond. Using van't Hoff plots, thermodynamic parameters for the pedal motion were determined. Computationally very cheap Atom–Atom-Force Field (AA-CLP) calculations were employed, which showed that the differences in dynamic disorder enthalpy between the two compounds are predominantly due to intermolecular interactions. AA-CLP calculations and gas phase electronic structure calculations were employed to show the link between intermolecular interactions and activation energy for pedal motion.
 Please wait while we load your content...
Please wait while we load your content...

