
The nature of the C–Cl⋯Cl–C intermolecular interactions found in molecular crystals: a general theoretical-database study covering the 2.75–4.0 Å range
Abstract
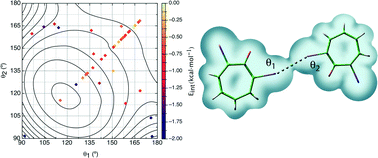
The nature of C–Cl⋯Cl–C interactions in molecular crystals has been evaluated at the MP2/aug-cc-pVDZ computational level after test computations on simple model systems showed that such a computational level predicts for model dimers the same angular dependence and impact of the C(ipso) atom hybridization as those at the MP2/CBS computational levels. Thus MP2/aug-cc-pVDZ calculations predict a C(spn)–Cl⋯Cl–C(spn) strength of −0.73, −0.87, and −0.96 kcal mol−1 for n = 3, n = 2 (non-aromatic), and n = 1 (all BSSE-corrected values) at their most stable orientation, while for the same orientations and hybridization MP2/CBS calculations predict values of −1.14, −1.29, and −1.40 kcal mol−1. The first group of computations on the model dimers allows conclusion that the strength of the C–Cl⋯Cl–C interactions depends on (a) the number of short-distance Cl⋯Cl contacts involved, (b) the hybridization of the C(ipso) atom [E(Csp2) > E(Csp) > E(Csp3)], (c) the degree of chlorination of the C(ipso) atom, and (d) the relative orientation of the two C–Cl groups. Two types of minima were found in the E(θ1, θ2) potential energy surface [θ1 = <C(1)–Cl(2)⋯Cl(3) and θ2 = <Cl(2)⋯Cl(3)–C(4)]: type I minima, where θ1 = θ2 = 90°, and type II minima, which is energetically more stable, where θ1 = 180° and θ2 = 90° or θ1 = 90° and θ2 = 180° (the orientation where θ1 = θ2 = 155° (a type I geometry) is a saddle point in E(θ1, θ2) that connects the two type II minima). The interaction energy was also computed for 45 C–Cl⋯Cl–C containing dimers extracted from the Cambridge Crystallographic Database (CCDB) and having a Cl⋯Cl distance smoothly distributed within the 2.75–4.0 Å range. The nature of these interactions was further characterized by looking at (a) the dominant component of the dimer interaction energy and (b) the characteristic properties of their only Cl⋯Cl bond critical point (evaluated from an atoms-in-molecules (AIM) analysis of the dimer electron density). Their interaction energy is dominated by the dispersion component, although a much weaker electrostatic component is also present in some cases. These interactions fail to fulfill the strength–length distribution that should correlate Eint and the Cl⋯Cl distance. A previously proposed correlation between the electron density at the Cl⋯Cl bond critical point and the strength of the C–Cl⋯Cl–C interaction is shown to fail for short Cl⋯Cl distances.
- This article is part of the themed collection: International Year of Crystallography Celebration: Europe and South Africa
 Please wait while we load your content...
Please wait while we load your content...

