
Dual deoxygenation in an α-ketoimine chelated rhenium(iii) complex: structural and mechanistic interpretations†

Abstract
An unprecedented case of dual deoxygenation is demonstrated in rhenium chemistry. It is authenticated that an oxorhenium(V) motif and a chelated diaryl-α-ketooxime ligand undergo concurrent oxygen atom transfer (OAT) to form a triarylphosphine oxide coordinated ReIII–α-ketoimine complex. The two OAT events are mutually dependent. OAT-induced ReV![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O → ReIII–OPR3 conversion must occur prior to the OAT-mediated α-ketooxime → α-ketoimine transformation. The first intramolecular OAT occurs across a free energy barrier of 29.1 kcal mol−1, and subjacent molecular orbital effects related to
O → ReIII–OPR3 conversion must occur prior to the OAT-mediated α-ketooxime → α-ketoimine transformation. The first intramolecular OAT occurs across a free energy barrier of 29.1 kcal mol−1, and subjacent molecular orbital effects related to 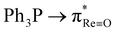 charge transfer are identified. The N–O bond cleavage of the oxime is induced by oxidative addition at the ReIII centre across a free energy barrier of 25.8 kcal mol−1 to afford a reactive ReV–hydroxo intermediate. The second intramolecular OAT involves electron transfer between the ReV-bound hydroxo and PPh3 moieties. Due to increased nucleophilicity of the hydroxo group, the second OAT is kinetically facile, with a low activation barrier of 8.3 kcal mol−1. Interestingly, while PPh3 acts as a nucleophile in the first OAT, it behaves as an electrophile in the second. Deoxygenation of diaryl-α-ketooxime is halted upon replacing the oxorhenium(V) motif by a kinetically nonlabile imidorhenium(V) moiety in the ReV–precursor. In that case, deprotonation of oxime occurs exclusively to generate the ReV–α-ketooximato complex. The predominance of the C-nitroso form of the oxime in the ReV–α-ketooximato species is a notable and hitherto unreported feature in rhenium chemistry. The aforementioned reactions of diaryl-α-ketooxime elegantly highlight ReV-substrate selectivity, which is justified through comprehensive mechanistic analysis.
charge transfer are identified. The N–O bond cleavage of the oxime is induced by oxidative addition at the ReIII centre across a free energy barrier of 25.8 kcal mol−1 to afford a reactive ReV–hydroxo intermediate. The second intramolecular OAT involves electron transfer between the ReV-bound hydroxo and PPh3 moieties. Due to increased nucleophilicity of the hydroxo group, the second OAT is kinetically facile, with a low activation barrier of 8.3 kcal mol−1. Interestingly, while PPh3 acts as a nucleophile in the first OAT, it behaves as an electrophile in the second. Deoxygenation of diaryl-α-ketooxime is halted upon replacing the oxorhenium(V) motif by a kinetically nonlabile imidorhenium(V) moiety in the ReV–precursor. In that case, deprotonation of oxime occurs exclusively to generate the ReV–α-ketooximato complex. The predominance of the C-nitroso form of the oxime in the ReV–α-ketooximato species is a notable and hitherto unreported feature in rhenium chemistry. The aforementioned reactions of diaryl-α-ketooxime elegantly highlight ReV-substrate selectivity, which is justified through comprehensive mechanistic analysis.

- This article is part of the themed collection: Celebrating International Women’s Day 2026: Women in Inorganic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

