
Bridging physical intuition and hardware efficiency for correlated electronic states: the local unitary cluster Jastrow ansatz for electronic structure†

Abstract
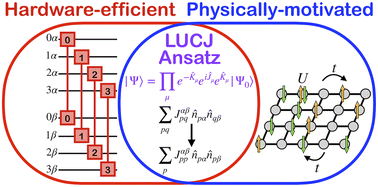
A prominent goal in quantum chemistry is to solve the molecular electronic structure problem for ground state energy with high accuracy. While classical quantum chemistry is a relatively mature field, the accurate and scalable prediction of strongly correlated states found, e.g., in bond breaking and polynuclear transition metal compounds remains an open problem. Within the context of a variational quantum eigensolver, we propose a new family of ansatzes which provides a more physically appropriate description of strongly correlated electrons than a unitary coupled cluster with single and double excitations (qUCCSD), with vastly reduced quantum resource requirements. Specifically, we present a set of local approximations to the unitary cluster Jastrow wavefunction motivated by Hubbard physics. As in the case of qUCCSD, exactly computing the energy scales factorially with system size on classical computers but polynomially on quantum devices. The local unitary cluster Jastrow ansatz removes the need for SWAP gates, can be tailored to arbitrary qubit topologies (e.g., square, hex, and heavy-hex), and is well-suited to take advantage of continuous sets of quantum gates recently realized on superconducting devices with tunable couplers. The proposed family of ansatzes demonstrates that hardware efficiency and physical transparency are not mutually exclusive; indeed, chemical and physical intuition regarding electron correlation can illuminate a useful path towards hardware-friendly quantum circuits.
 Please wait while we load your content...
Please wait while we load your content...

