
Radiometallation and photo-triggered release of ready-to-inject radiopharmaceuticals from the solid phase†
 a
Eszter
Boros
*a
a
Eszter
Boros
*a
Abstract
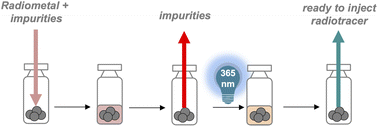
The efficient, large-scale synthesis of radiometallated radiopharmaceuticals represents an emerging clinical need which, to date, is inherently limited by time consuming, sequential procedures to conduct isotope separation, radiochemical labeling and purification prior to formulation for injection into the patient. In this work, we demonstrate that a solid-phase based, concerted separation and radiosynthesis strategy followed by photochemical release of radiotracer in biocompatible solvents can be employed to prepare ready-to-inject, clinical grade radiopharmaceuticals. Optimization of resin base, resin loading, and radiochemical labeling capacity are demonstrated with 67Ga and 64Cu radioisotopes using a short model peptide sequence and further validated using two peptide-based radiopharmaceuticals with clinical relevance, targeting the gastrin-releasing peptide and the prostate specific membrane antigen. We also demonstrate that the solid-phase approach enables separation of non-radioactive carrier ions Zn2+ and Ni2+ present at 105-fold excess over 67Ga and 64Cu by taking advantage of the superior Ga3+ and Cu2+ binding affinity of the solid-phase appended, chelator-functionalized peptide. Finally, a proof of concept radiolabeling and subsequent preclinical PET-CT study with the clinically employed positron emitter 68Ga successfully exemplifies that Solid Phase Radiometallation Photorelease (SPRP) allows the streamlined preparation of radiometallated radiopharmaceuticals by concerted, selective radiometal ion capture, radiolabeling and photorelease.
 Please wait while we load your content...
Please wait while we load your content...

