
A computational investigation towards substitution effects on 8π electrocyclisation of conjugated 1,3,5,7-octatetraenes†
 a
and
Nadeem S.
Sheikh
*a
a
and
Nadeem S.
Sheikh
*a
Abstract
A computational investigation using M06-2X/6-31+G(d) method is reported for the substitution effects on 8π electrocyclisation of conjugated octatetraene. This systematic study describes the mono- and di-substitution effect across the 1,3,5,7-octatetraene skeleton. A general preference of the outward substitution over the inward, at C1 position of the monosubstituted system is observed. However, mesomerically electron donating group (–NH2 and –OH) display an opposite effect with respect to secondary orbital interaction (SOI) between the lone pair on the substituent and the 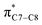 orbital. A comparative evaluation on the computed activation energies for the 1-, 2-, 3-, and 4-monosubstituted system showed an insignificant impact on the rate of the reaction, in contrast to the electrocyclic ring closure of the unsubstituted compound. Computations of disubstituted system are more pronounced, where a remarkable acceleration is observed for 2-NO2–7-NO2 substituted octatetraene at 4.9 kcal mol−1, and a noticeable deceleration for 4-CH3–5-CH3 substituted octatetraene at 25.4 kcal mol−1 from the parent molecule, 17.0 kcal mol−1. A visible accelerated effects are commonly exhibited by the substitution on the terminal double bonds (C1, C2, C7, and C8), that are 1,2-, 1,7-, 1,8-, and 2,7-patterns, in regard to the greater orbital interaction for the new σ-bond formation. Despite the unfavourable steric clashes of the substituents in the 1,8-system, an apparent reduction in the energy barrier up to 7.4 kcal mol−1 is computed for 1-NH2–8-NO2 system from 17.0 kcal mol−1. This is due to the synergistic effect of the electron donor and electron acceptor, enhancing the stability of the transition structure. The electrocyclic ring closure involving vicinal substitution patterns, such as 1,2-, 2,3-, 3,4-, and 4,5-systems are critically dominated by steric crowding between the adjacent functional groups. In certain cases of the 1,2-substituted system, a noticeable accelerated effects are found for 1-NH2–2-NH2-substituted compound (9.7 kcal mol−1) due to an increased in electronic density on the substituted terminal double bond (C1–C2), hence favouring the formation of the new σ-bond.
orbital. A comparative evaluation on the computed activation energies for the 1-, 2-, 3-, and 4-monosubstituted system showed an insignificant impact on the rate of the reaction, in contrast to the electrocyclic ring closure of the unsubstituted compound. Computations of disubstituted system are more pronounced, where a remarkable acceleration is observed for 2-NO2–7-NO2 substituted octatetraene at 4.9 kcal mol−1, and a noticeable deceleration for 4-CH3–5-CH3 substituted octatetraene at 25.4 kcal mol−1 from the parent molecule, 17.0 kcal mol−1. A visible accelerated effects are commonly exhibited by the substitution on the terminal double bonds (C1, C2, C7, and C8), that are 1,2-, 1,7-, 1,8-, and 2,7-patterns, in regard to the greater orbital interaction for the new σ-bond formation. Despite the unfavourable steric clashes of the substituents in the 1,8-system, an apparent reduction in the energy barrier up to 7.4 kcal mol−1 is computed for 1-NH2–8-NO2 system from 17.0 kcal mol−1. This is due to the synergistic effect of the electron donor and electron acceptor, enhancing the stability of the transition structure. The electrocyclic ring closure involving vicinal substitution patterns, such as 1,2-, 2,3-, 3,4-, and 4,5-systems are critically dominated by steric crowding between the adjacent functional groups. In certain cases of the 1,2-substituted system, a noticeable accelerated effects are found for 1-NH2–2-NH2-substituted compound (9.7 kcal mol−1) due to an increased in electronic density on the substituted terminal double bond (C1–C2), hence favouring the formation of the new σ-bond.

 Please wait while we load your content...
Please wait while we load your content...

