
IrI(η4-diene) precatalyst activation by strong bases: formation of an anionic IrIII tetrahydride†
 ab
Agustí
Lledós,
c
Adrian C.
Whitwood,
a
Jason M.
Lynam,
a
John M.
Slattery,
a
Simon B.
Duckett
*a
and
Rinaldo
Poli
*bd
ab
Agustí
Lledós,
c
Adrian C.
Whitwood,
a
Jason M.
Lynam,
a
John M.
Slattery,
a
Simon B.
Duckett
*a
and
Rinaldo
Poli
*bd
Abstract
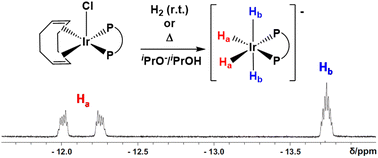
The reaction between [IrCl(COD)]2 and dppe in a 1 : 2 ratio was investigated in detail under three different conditions. [IrCl(COD)(dppe)], 1, is formed at room temperature in the absence of base. In the presence of a strong base at room temperature, hydride complexes that retain the carbocyclic ligand in the coordination sphere are generated. In isopropanol, 1 is converted into [IrH(1,2,5,6-η2:η2-COD)(dppe)] (2) on addition of KOtBu, with k12 = (1.11 ± 0.02) × 10−4 s−1, followed by reversible isomerisation to [IrH(1-κ-4,5,6-η3-C8H12)(dppe)] (3) with k23 = (3.4 ± 0.2) × 10−4 s−1 and k32 = (1.1 ± 0.3) × 10−5 s−1 to yield an equilibrium 5 : 95 mixture of 2 and 3. However, when no hydride source is present in the strong base (KOtBu in benzene or toluene), the COD ligand in 1 is deprotonated, followed by β-H elimination of an IrI–C8H11 intermediate, which leads to complex [IrH(1-κ-4,5,6-η3-C8H10)(dppe)] (4) selectively. This is followed by its reversible isomerisation to 5, which features a different relative orientation of the same ligands (k45 = (3.92 ± 0.11) × 10−4 s−1; k5-4 = (1.39 ± 0.12) × 10−4 s−1 in C6D6), to yield an equilibrated 32 : 68 mixture of 4 and 5. DFT calculations assisted in the full rationalization of the selectivity and mechanism of the reactions, yielding thermodynamic (equilibrium) and kinetic (isomerization barriers) parameters in excellent agreement with the experimental values. Finally, in the presence of KOtBu and isopropanol at 80 °C, 1 is transformed selectively to K[IrH4(dppe)] (6), a salt of an anionic tetrahydride complex of IrIII. This product is also selectively generated from 2, 3, 4 and 5 and H2 at room temperature, but only when a strong base is present. These results provide an insight into the catalytic action of [IrCl(COD)(LL)] complexes in the hydrogenation of polar substrates in the presence of a base.
 Please wait while we load your content...
Please wait while we load your content...

