
Revealing the incorporation of an NH2 group into the edge of carbon dots for H2O2 sensing via the C–N⋯H hydrogen bond interaction†
 *a
and
*a
and
Abstract
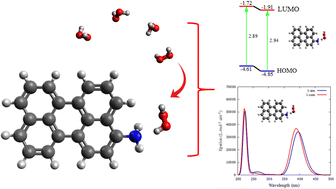
We investigated hydrogen peroxide (H2O2) sensing on NH2-functionalized carbon dots (Cdots) for three different –NH2 positions, and the N atom was found to be the active site using a quantum computational approach. B3LYP and 6-31G(d,p) were used for density functional theory (DFT) ground state calculations, whereas CAM-B3LYP and the same basis set were used in time-dependent density functional theory (TD-DFT) excited state calculations. Structural optimization showed that the H2O2 is chemisorbed on 1-sim via a C–N⋯H hydrogen bond interaction with an adsorption energy of −10.61 kcal mol−1. Mulliken atomic charge distributions and electrostatic potential (ESP) analysis were both used to determine reactivity of the molecules at the atomic level. For in-depth analysis of the ground states, we utilized Frontier molecular orbital (FMO) theory, quantum theory of atoms in molecules (QTAIM), and non-covalent interaction (NCI) index analysis. In addition, we also present UV-vis absorption spectra and charge transfer lengths to understand the mechanism of H2O2 sensing in excited states. Based on the molecular and electronic properties of the NH2-Cdots, it was shown that 1-sim is a potential candidate for use as an electrochemical sensor for H2O2 sensing. Whereas 3-sim is believed to be a potential candidate for use as an optical sensor of H2O2 based on the UV-vis characteristics via photoinduced charge transfer.
 Please wait while we load your content...
Please wait while we load your content...

