
Polymorphism of boron phosphide: theoretical investigation and experimental assessment
Vladimir L.
Solozhenko  *a
and
Samir F.
Matar
†b
*a
and
Samir F.
Matar
†b
*a
and
Samir F.
Matar
†b
Abstract
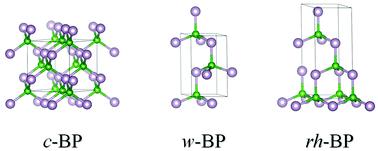
Stable crystal structures of wurtzite (w-BP) and recently discovered rhombohedral (rh-BP) polymorphic modifications of boron phosphide were obtained based on crystal chemistry rationale and unconstrained geometry optimization calculations within density functional theory (DFT), and compared with the known cubic polymorph (c-BP). Both w-BP and rh-BP are mechanically (elastic constants) and dynamically (phonons) stable and exhibit thermodynamic and mechanical properties very close to those of c-BP. The electronic band structures of all BP polymorphs exhibit semi-conducting behavior with band gap magnitudes close to 2 eV.
 Please wait while we load your content...
Please wait while we load your content...

