
Polymorphic self-assembly of helical tubules is kinetically controlled†

Abstract
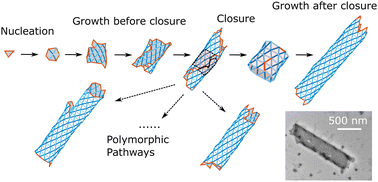
In contrast to most self-assembling synthetic materials, which undergo unbounded growth, many biological self-assembly processes are self-limited. That is, the assembled structures have one or more finite dimensions that are much larger than the size scale of the individual monomers. In many such cases, the finite dimension is selected by a preferred curvature of the monomers, which leads to self-closure of the assembly. In this article, we study an example class of self-closing assemblies: cylindrical tubules that assemble from triangular monomers. By combining kinetic Monte Carlo simulations, free energy calculations, and simple theoretical models, we show that a range of programmable size scales can be targeted by controlling the intricate balance between the preferred curvature of the monomers and their interaction strengths. However, their assembly is kinetically controlled—the tubule morphology is essentially fixed shortly after closure, resulting in a distribution of tubule widths that is significantly broader than the equilibrium distribution. We develop a simple kinetic model based on this observation and the underlying free-energy landscape of assembling tubules that quantitatively describes the distributions. Our results are consistent with recent experimental observations of tubule assembly from triangular DNA origami monomers. The modeling framework elucidates design principles for assembling self-limited structures from synthetic components, such as artificial microtubules that have a desired width and chirality.
 Please wait while we load your content...
Please wait while we load your content...

