
Unraveling the origin of reductive stability of super-concentrated electrolytes from first principles and unsupervised machine learning†
 *a
*a
Abstract
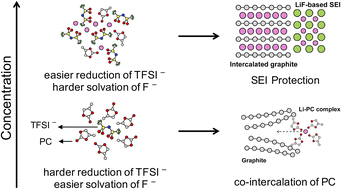
Developing electrolytes with excellent electrochemical stability is critical for next-generation rechargeable batteries. Super-concentrated electrolytes (SCEs) have attracted great interest due to their high electrochemical performances and stability. Previous studies have revealed changes in solvation structures and shifts in lowest unoccupied molecular orbitals from solvents to anions, promoting the formation of an anion-derived solid-electrolyte-interphase (SEI) in SCE. However, a direct connection at the atomic level to electrochemical properties is still missing, hindering the rational optimization of electrolytes. Herein, we combine ab initio molecular dynamics with the free energy calculation method to compute redox potentials of propylene carbonate electrolytes at a range of LiTFSI concentrations, and moreover employ an unsupervised machine learning model with a local structure descriptor to establish the structure–property relations. Our calculation indicates that the network of TFSI− in SCE not only helps stabilize the added electron and renders the anion more prone to reductive decomposition, but also impedes the solvation of F− and favors LiF precipitation, together leading to effective formation of protective SEI layers. Our work provides new insights into the solvation structures and electrochemistry of concentrated electrolytes which are essential to electrolyte design in batteries.
 Please wait while we load your content...
Please wait while we load your content...

