
Stereoretentive and regioselective selenium-catalyzed intermolecular propargylic C–H amination of alkynes†

Abstract
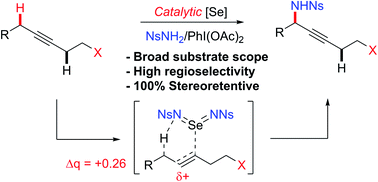
Herein we report an intermolecular propargylic C–H amination of alkynes. This reaction is operationally convenient and requires no transition metal catalysts or additives. Terminal, silyl, and internal alkynes bearing a wide range of functional groups can be aminated in high yields. The regioselectivity of amination for unsymmetrical internal alkynes is strongly influenced by substitution pattern (tertiary > secondary > primary) and by relatively remote heteroatomic substituents. We demonstrate that amination of alkynes bearing α-stereocenters occurs with retention of configuration at the newly-formed C–N bond. Competition experiments between alkynes, kinetic isotope effects, and DFT calculations are performed to confirm the mechanistic hypothesis that initial ene reaction of a selenium bis(imide) species is the rate- and product-determining step. This ene reaction has a transition state that results in substantial partial positive charge development at the carbon atom closer to the amination position. Inductive and/or hyperconjugative stabilization or destabilization of this positive charge explains the observed regioselectivities.
 Please wait while we load your content...
Please wait while we load your content...

