
Kinetics and mechanism of the Barton–Kellogg olefination: a computational DFT study using CTST theory and topological approaches†

Abstract
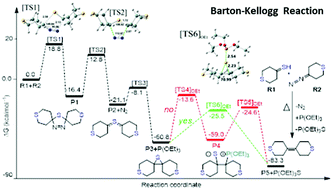
The kinetics and mechanism of the Barton–Kellogg olefination have been studied theoretically. In particular, the detailed mechanism of the reaction of tetrahydro-4H-thiopyran-4-thione (R1) and 4-diazotetrahydro-2H-thiopyran (R2) forming 2,2′,3,3′,5,5′,6,6′-octahydro-4,4′-bithiopyranylidene (P5) was investigated. The non-steady state approximation on the potential energy surface indicates that desulfurization of the episulfide (P3) by P(OEt)3 is the rate-determining step (RDS) and the obtained rate constant corrected by the Eckart tunneling model over the temperature range of 306.9–460.3 K can be expressed as k = 3.44 × 10−15exp (−21.09 kcal mol−1/RT). When PH3 is substituted for P(OEt)3 in the reaction, re-examination of the RDS showed that this structural simplification cannot model the reaction accurately. Topological analysis and synchronicity index indicate that the desulfurization of the episulfide by P(OEt)3 in toluene at a temperature of 383.6 K is a concerted process in a slightly asynchronous manner. The synergetic effects of the electron localization function (ELF), non-covalent interaction analysis (NCI), and quantum theory of atoms-in-molecules (QTAIM) techniques unraveled the molecular mechanism of this reaction. The chemical events along the reaction path are heterolytic rupture of C1,2–S and formation of P–S bonds, which can be described by the sequence of catastrophes 5-(EE) (FF)EC†-0.
 Please wait while we load your content...
Please wait while we load your content...

