
Thiol–yne chemistry of diferrocenylacetylene: from synthesis and electrochemistry to theoretical studies†
 *ab
Antonio
Valverde-González,
‡a
M. Merced
Montero-Campillo,
c
Otilia
Mó
bc
and
Isabel
Cuadrado
ab
*ab
Antonio
Valverde-González,
‡a
M. Merced
Montero-Campillo,
c
Otilia
Mó
bc
and
Isabel
Cuadrado
ab
Abstract
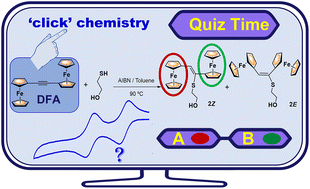
The thiol–yne coupling chemistry of diferrocenylacetylene (FcC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CFc) 1, bearing two electron rich and redox-active ferrocenyl units (Fc = Fe(η5-C5H4)(η5-C5H5)) and an internal triple bond, has been investigated for the first time. In order to determine whether steric limitations might affect hydrothiolation, a model reaction using a functionalized monothiol was tested, namely 2-mercaptoethanol I. The thiol–diferrocenylacetylene reactions were initiated either thermally (in toluene with AIBN) or by UV light irradiation (in THF and in the presence of DMPA as the photoinitiator). The outcomes of these thiol–yne reactions showed a strong dependence on the initiation method used, with the thermally initiated one being the most efficient. These thiol–diferrocenylacetylene reactions mainly afforded the (Z)-stereoisomer of the newly obtained vinyl thioether sulfide FcCH
CFc) 1, bearing two electron rich and redox-active ferrocenyl units (Fc = Fe(η5-C5H4)(η5-C5H5)) and an internal triple bond, has been investigated for the first time. In order to determine whether steric limitations might affect hydrothiolation, a model reaction using a functionalized monothiol was tested, namely 2-mercaptoethanol I. The thiol–diferrocenylacetylene reactions were initiated either thermally (in toluene with AIBN) or by UV light irradiation (in THF and in the presence of DMPA as the photoinitiator). The outcomes of these thiol–yne reactions showed a strong dependence on the initiation method used, with the thermally initiated one being the most efficient. These thiol–diferrocenylacetylene reactions mainly afforded the (Z)-stereoisomer of the newly obtained vinyl thioether sulfide FcCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Fc)S–(CH2)2OH (2), unlike the more common (E)-vinyl sulfides found in other additions to alkynes. The hydrothiolation of the internal –CC– bond in 1 was successfully extended to dithiol 2,2′-(ethylenedioxy)diethanethiol II, leading to the formation of the (ZZ)-isomer, with four ferrocenyl units, as the major product. According to the electrochemical studies, the new asymmetrical ferrocenyl–vinyl sulfides show iron–iron electronic and electrostatic interactions. Theoretical results for the (Z)-stereoisomer (2) suggest that adiabatic oxidation would lead to the loss of almost one electron on the ferrocenyl subunit closer to the thioether chain. Furthermore, the thiol–yne chemistry of the internal –CC– bond in diferrocenylacetylene has been compared to the external triple bond in ethynylferrocene, the theoretical results of which helped us to rationalize the very different reactivities observed in both metallocenes.
C(Fc)S–(CH2)2OH (2), unlike the more common (E)-vinyl sulfides found in other additions to alkynes. The hydrothiolation of the internal –CC– bond in 1 was successfully extended to dithiol 2,2′-(ethylenedioxy)diethanethiol II, leading to the formation of the (ZZ)-isomer, with four ferrocenyl units, as the major product. According to the electrochemical studies, the new asymmetrical ferrocenyl–vinyl sulfides show iron–iron electronic and electrostatic interactions. Theoretical results for the (Z)-stereoisomer (2) suggest that adiabatic oxidation would lead to the loss of almost one electron on the ferrocenyl subunit closer to the thioether chain. Furthermore, the thiol–yne chemistry of the internal –CC– bond in diferrocenylacetylene has been compared to the external triple bond in ethynylferrocene, the theoretical results of which helped us to rationalize the very different reactivities observed in both metallocenes.
 Please wait while we load your content...
Please wait while we load your content...

