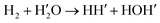An accurate full-dimensional H4O potential energy surface and dynamics of an exchange reaction
 †
†


Abstract
H2O and H2 are ubiquitous in the universe and their collisions play a crucial role in astrophysical processes. The reaction between the two also provides a prototype for understanding dynamics through four-center transition states. In this work, we adopted a strategy of combing two ab initio methods, CCSD(T)-F12a/AVTZ and MRCI-F12 + Qrot/AVTZ, to provide a balanced description for all regions of the configuration space, including one hydrogen exchange channel and two dissociation channels, namely H2 + H2O, H + H + H2O, and H + OH + H2. About 40 500 points were sampled to cover all dynamically relevant space, and the permutation invariant polynomial-neural network (PIP-NN) method was employed to develop a high-precision full-dimensional potential energy surface (PES), with a total fitting error of 0.055 kcal mol−1. Using a quasi-classical trajectory (QCT) method, the reaction dynamics and mode specificity of the hydrogen exchange channel were studied on the PES. It has been found that the stretching mode of H2, the bending, the symmetric stretching, the asymmetric stretching mode of H2O, and the translational mode can all promote reactivity. The strongest promotion effect comes from the H–H stretching mode. The Sudden Vector Projection (SVP) model was applied to predict mode specificity effects and rationalize the product energy partitioning. In both cases, the QCT and SVP results are generally consistent with each other. Furthermore, the hydrogen exchange channel was found to be dominated by sideways scattering.

 Please wait while we load your content...
Please wait while we load your content...