
Accurate computed singlet–triplet energy differences for cobalt systems: implication for two-state reactivity†

Abstract
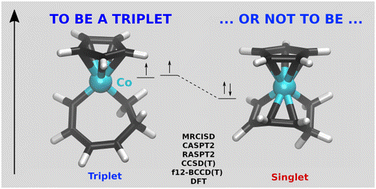
Accurate singlet–triplet energy differences for cobalt and rhodium complexes were calculated by using several wave function methods, such as MRCISD, CASPT2, CCSD(T) and BCCD(T). Relaxed energy differences were obtained by considering the singlet and triplet complexes, each at the minimum of their potential energy surfaces. Active spaces for multireference calculations were carefully checked to provide accurate results. The considered systems are built by increasing progressively the first coordination sphere around the metal. We included in our set two CpCoX complexes (Cp = cyclopentadienyl, X = alkenyl ligand), which have been suggested as intermediates in cycloaddition reactions. Indeed, cobalt systems have been used for more than a decade as active species in this kind of transformations, for which a two-state reactivity has been proposed. Most of the considered systems display a triplet ground state. However, in the case of a reaction intermediate, while a triplet ground state was predicted on the basis of Density Functional Theory results, our calculations suggest a singlet ground state. This stems from the competition between the exchange term (stabilising the triplet) and the accessibility of an intramolecular coordination (stabilising the singlet). This finding has an impact on the general mechanism of the cycloaddition reaction. Analogous rhodium systems were also studied and, as expected, they have a larger tendency to electron pairing than cobalt species.
 Please wait while we load your content...
Please wait while we load your content...

