
Donors, acceptors, and a bit of aromatics: electronic interactions of molecular adsorbates on hBN and MoS2 monolayers†
 ‡*ab
Juan Pablo
Guerrero-Felipe,
‡*cd
Ana M.
Valencia,
ce
Caterina
Cocchi
*ce
and
Marcella
Iannuzzi
*a
‡*ab
Juan Pablo
Guerrero-Felipe,
‡*cd
Ana M.
Valencia,
ce
Caterina
Cocchi
*ce
and
Marcella
Iannuzzi
*a
Abstract
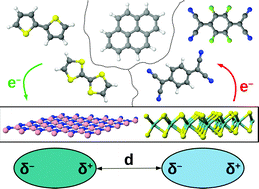
The design of low-dimensional organic–inorganic interfaces for the next generation of opto-electronic applications requires in-depth understanding of the microscopic mechanisms ruling electronic interactions in these systems. In this work, we present a first-principles study based on density-functional theory inspecting the structural, energetic, and electronic properties of five molecular donors and acceptors adsorbed on freestanding hexagonal boron nitride (hBN) and molybdenum disulfide (MoS2) monolayers. All considered interfaces are stable, due to the crucial contribution of dispersion interactions, which are maximized by the overall flat arrangement of the physisorbed molecules on both substrates. The level alignment of the hybrid systems depends on the characteristics of the constituents. On hBN, both type-I and type-II interfaces may form, depending on the relative energies of the frontier orbitals with respect to the vacuum level. On the other hand, all MoS2-based hybrid systems exhibit a type-II level alignment, with the molecular frontier orbitals positioned across the energy gap of the semiconductor. The electronic structure of the hybrid materials is further determined by the formation of interfacial dipole moments and by the wave-function hybridization between the organic and inorganic constituents. These results provide important indications for the design of novel low-dimensional hybrid materials with suitable characteristics for opto-electronics.
 Please wait while we load your content...
Please wait while we load your content...

