
CSinGe4−n2+ (n = 1–3): prospective systems containing planar tetracoordinate carbon (ptC)†

Abstract
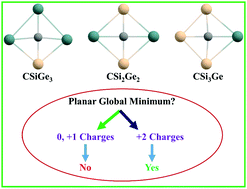
Density functional theory (DFT) based calculations have been carried out to explore the potential energy surface (PES) of CSinGe4−n2+/+/0 (n = 1–3) systems. The global minimum structures in the di-cationic states (1a, 1b, and 1c) contain a planar tetracoordinate carbon (ptC). For the CSi2Ge22+ system, the second stable isomer (2b) also contains a ptC with 0.67 kcal mol−1 higher energy than that of the 1b ptC isomer. The global minima of the neutral and mono-cationic states of the designed systems are not planar. The 1a, 1b, and 1c structures follow the 18 valence electron rule. The relative energies of the low-lying isomers of CSiGe32+, CSi2Ge22+, and CSi3Ge2+ systems with respect to the global minima were calculated using the CCSD(T)/aug-cc-pVTZ method. Ab initio molecular dynamics simulations for 50 ps time indicate that all the global minimum structures (1a, 1b, and 1c) are kinetically stable at 300 K and 500 K temperatures. The natural bond orbital (NBO) analysis suggests strong σ-acceptance of the ptC from the four surrounding atoms and simultaneously π-donation occurs from the ptC center. The nucleus independent chemical shift (NICS) showed σ/π-dual aromaticity. We hope that the designed di-cationic systems may be viable in the gas phase.
 Please wait while we load your content...
Please wait while we load your content...

