
A simple fragment-based method for van der Waals corrections over density functional theory†

Abstract
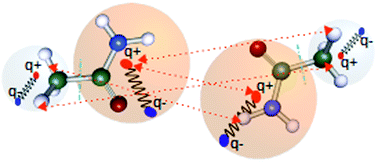
Modeling intermolecular noncovalent interactions between large molecules remains a challenge for the electron structure theory community due to the high cost. Fragment-based methods usually fare well in reducing the cost of computations in such systems. On the other hand, the interactions between quantum mechanical fluctuations of electron density can be modeled well by the interaction between atom-centered quantum Drude oscillators. In this paper, we have developed a simple yet effective method to describe intermolecular van der Waals interactions based on an amalgamation of Drude oscillator with the fragmentation of molecular systems. The resulting interaction energies have been used as corrections over low-cost DFT functional PBE. We have tested our method on the S66X8 database with significant success. While this work is a proof-of-concept study to employ anisotropic oscillators for modeling electron fluctuation, we have also reduced the number of empirical parameters compared to DFT-D methods without any loss of accuracy in the process. The resulting method shows accuracy at par with Grimme's DFT-D3 method while only using dipole–dipole interactions.
 Please wait while we load your content...
Please wait while we load your content...

