
The synergetic effect of an aqua ligand and metal site on the performance of single-atom catalysts in H2O2 synthesis: a density functional theory study†

Abstract
Studying the effect of the coordination field on the catalytic property is critical for the rational design of outstanding electrocatalysts for H2O2 synthesis. Herein, via density functional theory (DFT) calculations and ab initio molecular dynamic (AIMD) simulations, we built an effective computational framework to identify the synergetic effect of an aqua ligand and metal ion on the 2e− ORR catalytic performance under gas condition and aqua solvent. Specifically, the screening results of 29 single-atom catalysts (SACs), TM@C6N6 (TM = transition metal), indicated that Cu@C6N6 features excellent catalytic property with thermal stability, lowest 2e− ORR overpotential (0.02 V) and high selectivity of 99.99%. Once an aqua ligand binds with the Cu site, the activity is reduced to the overpotential of 0.42 V and the selectivity decreased slightly (99.98%) due to the reduction of the adsorption strength for the reaction intermediates. A combination of geometric structures and electronic properties revealed that such changes are correlated with the charge of the Cu site. Furthermore, based on molecular orbital theory, the essence of the high catalytic property deeply lies in the effect of the moderate electron back donation bond (dyz & dxz→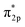 ) between Cu and O2. This work will provide a route to better design high-performance SACs for H2O2 synthesis effectively.
) between Cu and O2. This work will provide a route to better design high-performance SACs for H2O2 synthesis effectively.

 Please wait while we load your content...
Please wait while we load your content...

