
Intrachain photophysics of a donor–acceptor copolymer†
 *c
and
Oh-Hoon
Kwon
*ab
*c
and
Oh-Hoon
Kwon
*ab
Abstract
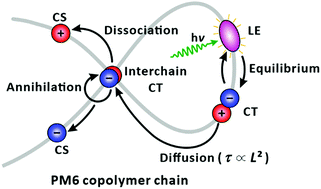
By taking advantage of bulk-heterojunction structures formed by blending conjugated donor polymers and non-fullerene acceptors, organic photovoltaic devices have recently attained promising power conversion efficiencies of above 18%. For optimizing organic photovoltaic devices, it is essential to understand the elementary processes that constitute light harvesters. Utilising femtosecond-resolved spectroscopic techniques that can access the timescales of locally excited (LE) state and charge-transfer (CT)/-separated (CS) states, herein we explored their photophysics in single chains of the top-notch performance donor–acceptor polymer, PM6, which has been widely used as a donor in state-of-the-art non-fullerene organic photovoltaic devices, in a single LE state per chain regime. Our observations revealed the ultrafast formation of a CT state and its equilibrium with the parent LE state. From the chain-length dependence of their lifetimes, the equilibrated states were found to idle until they reach a chain folding. At the chain folding, the CT state transforms into an interchain CT state that bifurcates into forming a CS state or annihilation within a picosecond. The observation of prevalent nonexponential behaviour in the relaxation of the transient species is attributed to the wide chain-length distribution that determines the emergence of the chain foldings in a single chain, thus, the lifetime of a LE and equilibrated CT states. Our findings indicate that the abundance of chain folding, where the generation of the “reactive” CS state is initiated from the interchain CT state, is essential for maximising charge carriers in organic photovoltaic devices based on PM6.
- This article is part of the themed collection: Developments in Ultrafast Spectroscopy
 Please wait while we load your content...
Please wait while we load your content...

