
Thermal properties of single-layer MoS2–WS2 alloys enabled by machine-learned interatomic potentials†

Abstract
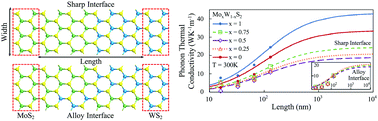
Two-dimensional (2D) quantum materials are poised to transform conventional electronics for a wide spectrum of applications that will encompass chemical sciences. For the study of thermal transport in single-layer (1L) or multi-layer transition metal dichalcogenides (TMDs), this work explores the combination of density functional theory (DFT) and algorithmic training for the generation of a moment tensor potential (MTP) that models 1L-MoS2, 1L-WS2 and their alloys, and demonstrates a synergy of theoretical techniques that is anticipated to play an important role in the field. From a high-performance computing perspective, these yield very convenient inter-atomic (or inter-molecular in other contexts) potentials that are useful to predict the response of quantum materials to thermal perturbations, or other driving forces. We show that our trained MTP functions successfully describe vibrational properties of the systems, and their thermal conductivities. The trained potential displays consistent agreement with DFT calculations, as well as the Stillinger–Weber (SW) potential. We also find that the thermal conductivity of the 2D alloys is little affected by sulfur vacancies. This is a behavior that may aid the fine-tuning of material's thermal properties for heat management and energy storage and conversion applications.
- This article is part of the themed collection: ChemComm Milestones – First Independent Articles
 Please wait while we load your content...
Please wait while we load your content...

