
Kinetic modelling of Pt/γ-Al2O3–Cl catalysts formulation changes in n-heptane reforming†
 *a
David
Farrusseng
b
and
*a
David
Farrusseng
b
and
Abstract
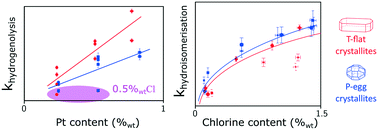
Bridging the gap between kinetic model conception and catalyst design is targeted in catalytic naphtha reforming process development. New catalysts are continuously optimised in order to achieve higher selectivity in C5+ products. An adequate description of catalytic transformations by the kinetic models would therefore provide clues for catalyst design and accelerate the time to market implementation of process simulators. This study investigates the influence of site density and location on n-heptane reforming selectivity. It identifies the nature of the limiting steps for the different reforming pathways on a broad range of catalyst formulations. A common lumped model using power law kinetics is developed to describe already published experimental observations on the set of selected catalysts. Linear free energy relationships are used in order to handle a reduced number of statistically relevant adjustable parameters. The dependence between reference rate constants and active phase formulation is then unravelled. Unexpected results indicate that chlorine content and repartition at the crystallite scale affects the hydrogenolysis activity. Within the range of tested formulations, this study suggests that hydroisomerisation reactions are limited by acid sites transformations whereas the aromatisation pathways seem to proceed through a metal/acid bi-functional scheme. The further elaboration of a kinetic model that is able to predict the effect of an industrial catalyst active phase formulation change in full naphtha cut reforming lies beyond the scope of this article.
 Please wait while we load your content...
Please wait while we load your content...

