
How aromatic system size affects the sensitivities of highly energetic molecules?†

Abstract
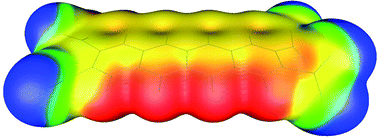
Positive values of electrostatic potentials above the central regions of the molecular surface are strongly related to the high sensitivities of highly energetic molecules. The influence of aromatic system size on the positive values of electrostatic potentials and bond dissociation energies of C–NO2 bonds was studied by Density Functional Theory (DFT) calculations on a series of polycyclic nitroaromatic molecules. Calculations performed at PBE/6-311G** level showed that with the increase of the aromatic system size, values of positive electrostatic potential above the central areas of selected energetic molecules decrease from 32.78 kcal mol−1 (1,2,4,5-tetranitrobenzene) to 15.28 kcal mol−1 (2,3,9,10-tetranitropentacene) leading to the decrease in the sensitivities of these molecules towards detonation. Results of the analysis of electrostatic potential maps were in agreement with the trends in bond dissociation energies calculated for C–NO2 bonds of studied nitroaromatic molecules. Bond dissociation energies values indicate that the C–NO2 bond in the molecule of 1,2,4,5-tetranitrobenzene (56.72 kcal mol−1) is weaker compared to the nitroaromatic molecules with the additional condensed aromatic rings and with a similar arrangement of –NO2 groups (59.75 kcal mol−1 in the case of 2,3,9,10-tetranitropentacene). The influence of the mutual arrangement of –NO2 groups on the sensitivity of nitroaromatic molecules was also analyzed. Results obtained within this study could be of great importance for the development of new classes of highly energetic molecules with lower sensitivity towards detonation.
- This article is part of the themed collection: #RSCPoster Conference
 Please wait while we load your content...
Please wait while we load your content...

