
Iridium-catalyzed highly stereoselective deoxygenation of tertiary cycloalkanols: stereoelectronic insights and synthetic applications†

Abstract
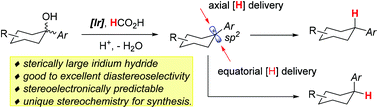
Excellent and unique diastereoselectivity is observed in the iridium-catalyzed deoxygenation of tertiary cyclohexanols and cyclopentanols. The substituent effect on the diastereoselectivity and detailed control models are analyzed case by case, using tertiary monocyclic and polycyclic cyclohexanols, bicyclic bridged cycloalkanols, and cyclopentanols as the model substrates. The selectivity is decided by the steric environment of the carbocation intermediates and is independent of the catalyst loading. Stereoelectronically, the iridium hydride approaches the carbocation in directions perpendicular to the carbocation plane. The sterically large iridium hydride delivers its hydride in the sterically least hindered direction to the carbocation. The deoxygenation has found important applications in the stereospecific arylations of sterically complex compounds. Our deoxygenation is stereochemically very different from the coupling reactions and can be used to specifically synthesize stereoisomers that are not available via cross-couplings.
- This article is part of the themed collection: Synthetic methodology in OBC
 Please wait while we load your content...
Please wait while we load your content...

