
On the role of steric and exchange–correlation effects in halogenated complexes
 *a
and
*a
and
Abstract
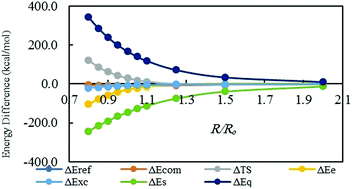
In this work, two formalisms of energy partitioning schemes in density functional theory (DFT) are utilized to find out what energetic components govern the origin of different types of interactions in halogenated complexes. The components in one of the energy partitioning schemes are the noninteracting kinetic, electrostatic, and exchange–correlation energies, while in the other approach the total electronic energy is decomposed into three independent components – steric, electrostatic, and fermionic quantum energies. Different sets of complexes in which halogens participate in a variety of interaction types at equilibrium geometries as well as along the potential energy curves have been considered as working models. With more or less different roles of different energetic terms, we find that besides the previously found important factors in this context, such as electrostatic and dispersion effects, the steric and exchange–correlation effects are the dominant factors contributing to the total interaction energies. Moreover, the obtained profiles of the energetic components along the potential energy curves reveal that the exchange–correlation effects, followed by the noninteracting kinetic energy and electrostatic component, are determinant contributions following the trend of interaction energies. Based on the reasonable and meaningful relationships between the interaction energies and their components for both the equilibrium and nonequilibrium geometries of the studied complexes, we also propose an alternative DFT energy partitioning scheme for investigation of noncovalent halogenated complexes, where the roles of the exchange–correlation, steric, and electrostatic effects are showcased.
 Please wait while we load your content...
Please wait while we load your content...

