
Halide-driven formation of lead halide perovskites: insight from ab initio molecular dynamics simulations†

Abstract
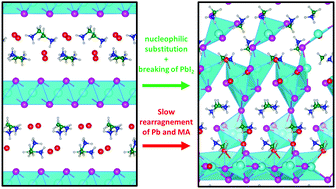
Controlling the crystallization mechanism of metal halide perovskites is of utmost importance to grow defect-less perovskite layers for efficient solar cells and optoelectronic devices. Despite its relevance, there is a lack of microscopic understanding of the nucleation and crystallization processes during the formation of the perovskite phase from its precursors. To unveil the electronic and atomistic features of this process we carry out ab initio molecular dynamics simulations on a model system which consists of a stoichiometric layered lead iodide (PbI2)·methylammonium iodide (MAI) structure, characteristic of intermediate phases observed in sequential deposition methods. Our results show clear evidence of halide-driven chemistry: MAI iodine ions attack lead ions in the PbI2 layers and cause a nucleophilic substitution of Pb–I bonds with a subsequent breaking of the PbI2 layer. Undercoordinated [PbIn]2−n complexes are initially formed which create the 3D perovskite framework mediated by additional nucleophilic attacks. The relatively fast rearrangement of [PbIn]2−n complexes followed by motion of MA cations limits the perovskite growth. Our results provide insight into the key steps of the perovskite formation on a microscopic scale, providing hitherto inaccessible details on the factors limiting the perovskite growth and on the effect of different halides on the kinetics of crystal formation.
- This article is part of the themed collection: #RSCPoster Conference
 Please wait while we load your content...
Please wait while we load your content...

