
Synthesis and coordination behaviour of aluminate-based quinolyl ligands†
 b
Andrew D.
Bond,
a
Raul
García-Rodríguez,
c
Dominic S.
Wright
*a
and
Annie L.
Colebatch
*d
b
Andrew D.
Bond,
a
Raul
García-Rodríguez,
c
Dominic S.
Wright
*a
and
Annie L.
Colebatch
*d
Abstract
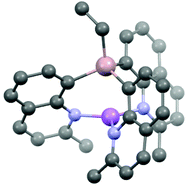
The effects of moving the donor N-atom from the 2-position in lithium (2-pyridyl)- and (2-quinolyl)aluminates to the more remote position in (8-quinolyl)aluminates have been investigated by solid-state structural and DFT computational studies of the new complexes [{EtAl(2-qy)3}Li(μ-X)Li(THF)3] (X = Cl/Br 62 : 38) [(1)Li(μ-X)Li(THF)3], [{(EtAl(2-qy)3)Li}2(μ-Br)]−Li(THF)4+ [{1Li}2(μ-Br)]−Li(THF)4+, [{EtAl(2-Me-8-qy)3}Li] [(2)Li], [{Me2Al(2-Me-8-qy)2}Li(THF)] [(3a)Li(THF)], [{Me2Al(6-Me-2-py)2}Li(THF)2] [(4)Li(THF)2] and [{{EtAl(2-Me-8-qy)2}2O}(Li2THF)] (5). Increasing the remoteness of the donor N-atom from the bridgehead results in large differences in the coordination of the Li+ cations by the (8-quinolyl)aluminate anions compared to 2-quinolyl or 2-pyridyl counterparts. The results are of potential interest in understanding how the coordination sites of ligands of this type can be tuned for the coordination requirements of specific metal centres.
 Please wait while we load your content...
Please wait while we load your content...

