
Oxygen-evolution reactions (OER) on transition-metal-doped Fe3Co(PO4)4 iron-phosphate surfaces: a first-principles study†

Abstract
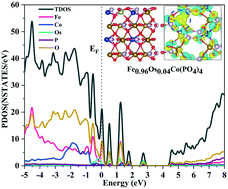
A series of transition-metal-doped Fe1−xMxCo(PO4)4(010) and Fe3Co1−xMx(PO4)4(010) electro-catalyst surfaces (with M = Mn, Os, Ru, Rh and Ir) have been modelled via density-functional theory (DFT) to gauge their mechanistic and thermodynamic prospects for oxygen-evolution reactions (OER), as part of the overall electrochemical water-splitting process. These doped surfaces have been analysed systematically by DFT calculations, scrutinising their underlying electronic- and nuclear-structure characteristics and thermodynamic-energetics profiles, to assess their OER viability, including consideration and comparison of charge-density effects for the transition-metal dopants. In particular, all of the reaction steps for the overall OER reaction schemes have been explored on all possible surface reaction sites, and the energy profile and associated free-energy-change effects have been computed and discussed. Iron replaced by osmium shows a most promising catalysing effect for all reactions at Co surface sites.
 Please wait while we load your content...
Please wait while we load your content...

