
Theoretical study of the stability, structure, and optical spectra of small silver clusters and their formation using density functional theory†

Abstract
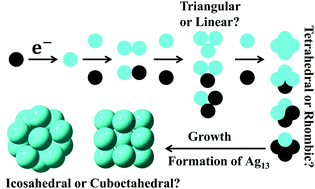
An understanding of the mechanism of formation of small clusters would help to identify efficient routes to their synthesis. Here, we apply density functional theory (DFT) to study the free energies and structure of ultra-small silver clusters, and time-dependent density functional theory (TDDFT) to calculate their UV-Vis spectra and provide a better understanding of the intermediate steps in cluster formation in the gas phase and solution. Our calculations of the optical properties of neutral and cationic clusters confirm the presence of charged and uncharged intermediates observed in pulse radiolysis experiments during the early stages of the growth of silver clusters. The free energies of formation of hydrated clusters extracted from DFT calculations reveal the greater thermodynamic stability of cationic clusters compared to the corresponding neutral clusters of the same composition. This is consistent with the predominance of kinetically stable cationic clusters observed in pulse radiolysis experiments. Our DFT and TDDFT calculations clarify the effects of ligand, hydration, and oxidation states on the structure, stability, and optical properties of silver clusters that elucidate the mechanism of silver cluster formation in solution and the gas phase.
 Please wait while we load your content...
Please wait while we load your content...

