
A comparative DFT study of the oxidation of Al crystals and nanoparticles†
 *a
*a
Abstract
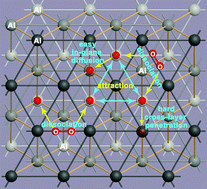
The thermodynamic and kinetic behaviors of O atoms on and in different Al nanoparticles (ANPs) and Al crystals have been systematically studied using first-principles calculations. The O adsorption strength on clean Al surfaces follows the order of (111) > ANPs > (110) > (100). The O adsorption strength on ANPs approaches that on Al(111) as the cluster size increases. The three-fold fcc-like sites on ANPs rather than the corner and edge sites are more favored by O* adatoms, which is due to the larger deformation energy related to the geometry change of ANPs when O is adsorbed at the corner and edge sites. The O adsorption behaviors on ANPs are different both from previous studies based on reactive force fields (ReaxFFs) and from those on transition metal clusters. The effective O–O interaction is short ranged (<5 Å), isotropic (in-plane and across-layer) and attractive over different Al surfaces and subsurfaces. The attraction is always about −0.1 eV per O pair at the first nearest neighboring (1NN) sites unless there is evident surface curvature or restructure. Due to the universal attraction, the O* adatoms either on the surface or in the subsurfaces prefer to form islands. In addition, any O diffusion away from the O islands will experience a much higher energy barrier than on a clear (sub)surface. Besides the most stable (Al2O3)n fragments, the metastable Al oxidation fragments are AlO4 monomers and (AlO2)n oligomers, which may be formed during intense oxidation and all feature tetra-coordinated Al and bi-coordinated O atoms.
 Please wait while we load your content...
Please wait while we load your content...

