
Adsorption and reaction mechanisms of single and double H2O molecules on graphene surfaces with defects: a density functional theory study†

Abstract
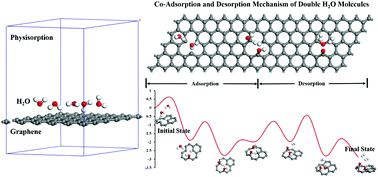
More attention needs to be drawn to the high application value of the gasification reaction between carbonaceous materials and water in industry. In this study, density functional theory is used to investigate the adsorption and reaction mechanism of water molecules on graphene surfaces with various kinds of defects. The desorption mechanism of the reaction product is also analyzed. The optimal and stable physical adsorption configuration of water molecules on the pristine graphene and various defects graphene surface has been determined. Chemisorption configurations of a single water molecule and double water molecules on the graphene surface with single vacancy defects are discussed and used as reaction precursors to explore the reaction path of water molecules in the process of desorbing hydrogen at active sites. The whole process of the reaction is largely exothermic and the thermodynamic advantages of double water molecules participating in the reaction are determined. The two reaction mechanisms of two-steps or co-adsorption and desorption of double water molecules are compared, and the lowest energy barrier advantage of the latter is determined.
 Please wait while we load your content...
Please wait while we load your content...

