
Theoretical study of the Cl(2P) + SiH4 reaction: global potential energy surface and product pair-correlated distributions. Comparison with experiment
 *a
and
J. C.
Corchado
a
*a
and
J. C.
Corchado
a
Abstract
For the theoretical study of the title reaction, an analytical full-dimensional potential energy surface named PES-2021 was developed for the first time, by fitting high-level explicitly-correlated ab initio data. This reaction presented high exothermicity, 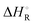 (298 K) = −11.6 kcal mol−1, reproducing the experimental evidence; it is a barrierless reaction and no intermediate complexes were found. PES-2021 is a continuous and smooth potential energy surface, it includes intuitive concepts in its development and fitting, such as stretching and bending nuclear motions, and it presents analytical first energy derivatives. Based on PES-2021, kinetics and dynamics studies were carried out using quasi-classical trajectory calculations. In the kinetics study, over the temperature range 300–450 K, we observed that rate constants were practically independent of temperature, with an almost zero activation energy, as compared to 0.0 and −0.48 kcal mol−1 experimentally reported. In this kinetics study the role of the spin–orbit effect on reactivity was analysed. In the dynamics study, different product pair-correlated dynamics properties were compared with the only experimental evidence: product energy partition, product vibrational distribution, product angular distribution and product speed distribution. We observed two mechanisms of reaction, a stripping mechanism associated with large impact parameters and forward scattering, and an indirect mechanism associated with sideways-backward scattering related with “nearly-trapped” trajectories due to the product rotation. In general, theoretical results reasonably simulate the experimental measurements when they consider some rotational and vibrational constraints as well as binning techniques to mimic a quantum-mechanical behaviour. Although the agreement is not quantitative, the present results shed light on the mechanism of this difficult polyatomic reactive system.
(298 K) = −11.6 kcal mol−1, reproducing the experimental evidence; it is a barrierless reaction and no intermediate complexes were found. PES-2021 is a continuous and smooth potential energy surface, it includes intuitive concepts in its development and fitting, such as stretching and bending nuclear motions, and it presents analytical first energy derivatives. Based on PES-2021, kinetics and dynamics studies were carried out using quasi-classical trajectory calculations. In the kinetics study, over the temperature range 300–450 K, we observed that rate constants were practically independent of temperature, with an almost zero activation energy, as compared to 0.0 and −0.48 kcal mol−1 experimentally reported. In this kinetics study the role of the spin–orbit effect on reactivity was analysed. In the dynamics study, different product pair-correlated dynamics properties were compared with the only experimental evidence: product energy partition, product vibrational distribution, product angular distribution and product speed distribution. We observed two mechanisms of reaction, a stripping mechanism associated with large impact parameters and forward scattering, and an indirect mechanism associated with sideways-backward scattering related with “nearly-trapped” trajectories due to the product rotation. In general, theoretical results reasonably simulate the experimental measurements when they consider some rotational and vibrational constraints as well as binning techniques to mimic a quantum-mechanical behaviour. Although the agreement is not quantitative, the present results shed light on the mechanism of this difficult polyatomic reactive system.

 Please wait while we load your content...
Please wait while we load your content...

