
A quantitative multiscale perspective on primary olefin formation from methanol

Abstract
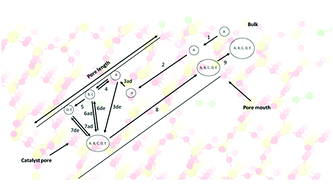
The formation of the first C–C bond and primary olefins from methanol over zeolite and zeotype catalysts has been studied for over 40 years. Over 20 mechanisms have been proposed for the formation of the first C–C bond. In this quantitative multiscale perspective, we decouple the adsorption, desorption, mobility, and surface reactions of early species through a combination of vacuum and sub-vacuum studies using temporal analysis of products (TAP) reactor systems, and through studies with atmospheric fixed bed reactors. These results are supplemented with density functional theory calculations and data-driven physical models, using partial differential equations, that describe the temporal and spatial evolution of species. We consider the effects of steam, early degradation species, and product masking due to the inherent autocatalytic nature of the process, which all complicate the observation of the primary olefin(s). Although quantitative spectroscopic determination of the lifetimes, surface mobility, and reactivity of adspecies is still lacking in the literature, we observe that reaction barriers are competitive with adsorption enthalpies and/or activation energies of desorption, while facile diffusion occurs in the porous structures of the zeolite/zeotype catalysts. Understanding the various processes allows for quantitative evaluation of their competing energetics, which leads to molecular insights as to what governs the catalytic activity during the conversion of methanol to primary olefins over zeolite/zeotype catalysts.
- This article is part of the themed collection: PCCP Perspectives
 Please wait while we load your content...
Please wait while we load your content...

