
Molecular dynamics investigation on the interaction of human angiotensin-converting enzyme with tetrapeptide inhibitors†
 a
Weihong
Min
*a
and
a
Weihong
Min
*a
and
Abstract
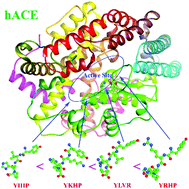
Angiotensin-converting enzyme (ACE) is a well-known zinc metalloenzyme whose physiological functions are vital to blood pressure regulation and management of hypertension. The development of more efficient peptide inhibitors is of great significance for the prevention and treatment of hypertension. In this research, molecular dynamics (MD) simulations were implemented to study the specific binding mechanism and interaction between human ACE (hACE) and tetrapeptides, YIHP, YKHP, YLVR, and YRHP. The calculation of relative binding free energy on the one hand verified that YLVR, an experimentally identified inhibitor, has a stronger inhibitory effect and, on the other hand, indicated that YRHP is the “best” inhibitor with the strongest binding affinity. Inspection of atomic interactions discriminated the specific binding mode of each tetrapeptide inhibitor with hACE and explained the difference of their affinity. Moreover, in-depth analysis of the MD production trajectories, including clustering, principal component analysis, and dynamic network analysis, determined the dynamic correlation between tetrapeptides and hACE and obtained the communities’ distribution of a protein–ligand complex. The present study provides essential insights into the binding mode and interaction mechanism of the hACE–peptide complex, which paves a path for designing effective anti-hypertensive peptides.
 Please wait while we load your content...
Please wait while we load your content...

