
Hydrogen abstraction/addition reactions in soot surface growth†

Abstract
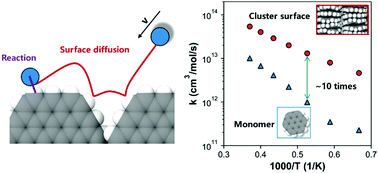
The hydrogen abstraction (HB) and addition reactions (HD) by H radicals are examined on a series of polycyclic aromatic hydrocarbon (PAH) monomers and models of quasi-surfaces using quasi-classical trajectory (QCT) method. QCT results reproduce the rate constants of HB reactions on PAH monomers from density functional theory (DFT) in the range of 1500–2700 K. The PAH size has a minor impact on the rates of HB reactions, especially at temperatures beyond 2100 K. In contrast, HD reactions have a clear size dependence, and a larger PAH yields a higher rate. It was also found that the preferred reaction pathway changes from HB to HD reactions at ∼1900 K. The rates of surface HB and HD reactions exceed those in the gas phase by nearly one factor of magnitude. Further analysis of the detailed trajectory of the QCT method reveals that about 50% of surface reactions can be attributed to the events of surface diffusion, which depends on the local energy transfer in gas-surface interactions. However, this phenomenon is not preferred in PAH monomers, as expected. Our finding here questions the treatment of the surface reactions of soot as the product of the first collision between the gaseous species and particle surface. The surface diffusion-induced reactions should be accounted for in the rates of the surface HB and HD reactions. The rate constants of HB and HD reactions on each reactive site (surface zig-zag, surface free-edge and pocket free-edge sites) were calculated by QCT method, and are recommended for the further development of surface chemistry models in soot formation.
 Please wait while we load your content...
Please wait while we load your content...

