
Theoretical description of molecular permeation via surface diffusion through graphene nanopores
 *a
Runfeng
Zhou
a
and
*a
Runfeng
Zhou
a
and
Abstract
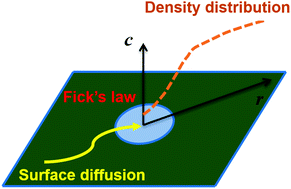
We establish a theoretical model to describe the surface molecular permeation through two-dimensional graphene nanopores based on the surface diffusion equation and Fick's law. The model is established by considering molecular adsorption and desorption from the surface adsorption layer and the molecular diffusion and concentration gradient on the graphene surface. By comparing with the surface flux obtained from molecular dynamics simulations, it is shown that the model can predict well the overall permeation flux especially for strongly adsorbed molecules (i.e. CO2 and H2S) on graphene surfaces. Although good agreement between the theoretical and simulated density distribution is hard to achieve owing to the large uncertainty in the calculation of surface diffusion coefficients based on the Einstein equation, the model itself is very competent to describe the surface molecular permeation both from the aspects of the overall permeation flux and detailed density distribution. This model is believed to supplement the theoretical description of molecular permeation through graphene nanopores and provide a good reference for the description of mass transport through two-dimensional porous materials.
 Please wait while we load your content...
Please wait while we load your content...

