
Cyclic peptides: backbone rigidification and capability of mimicking motifs at protein–protein interfaces†

Abstract
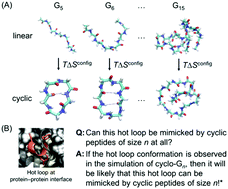
Cyclization is commonly employed in efforts to improve the target binding affinity of peptide-based probes and therapeutics. Many structural motifs have been identified at protein–protein interfaces and provide promising targets for inhibitor design using cyclic peptides. Cyclized peptides are generally assumed to be rigidified relative to their linear counterparts. This rigidification potentially pre-organizes the molecules to interact properly with their targets. However, the actual impact of cyclization on, for example, peptide configurational entropy, is currently poorly understood in terms of both its magnitude and molecular-level origins. Moreover, even with thousands of desired structural motifs at hand, it is currently not possible to a priori identify the ones that are most promising to mimic using cyclic peptides nor to select the ideal linker length. Instead, labor-intensive chemical synthesis and experimental characterization of various cyclic peptide designs are required, in hopes of finding one with improved target affinity. Herein, using molecular dynamics simulations of polyglycines, we elucidated how head-to-tail cyclization impacts peptide backbone dihedral entropy and developed a simple strategy to rapidly screen for structures that can be reliably mimicked by preorganized cyclic peptides. As expected, cyclization generally led to a reduction in backbone dihedral entropy; notably, however, this effect was minimal when the length of polyglycines was >9 residues. We also found that the reduction in backbone dihedral entropy upon cyclization of small polyglycine peptides does not result from more restricted distributions of the dihedrals; rather, it was the correlations between specific dihedrals that caused the decrease in configurational entropy in the cyclic peptides. Using our comprehensive cyclo-Gn structural ensembles, we obtained a holistic picture of what conformations are accessible to cyclic peptides. Using “hot loops” recently identified at protein–protein interfaces as an example, we provide clear guidelines for choosing the “easiest” hot loops for cyclic peptides to mimic and for identifying appropriate cyclic peptide lengths. In conclusion, our results provide an understanding of the thermodynamics and structures of this interesting class of molecules. This information should prove particularly useful for designing cyclic peptide inhibitors of protein–protein interactions.
 Please wait while we load your content...
Please wait while we load your content...

